原来,历时一个月,小T哥远赴宁夏、山东、河北、广东、浙江和天津多地,对近20家单位进行了宣讲和交流。内容覆盖了微生物&代谢组联合分析、细胞STR鉴定、SNP/SSR分子标记、全转录组联合分析和DNA甲基化检测等多个领域,受到宣讲单位老师的一致好评。再次感谢支持阅微基因巡讲活动的单位和老师们,您们的支持和指导让我们越来越强大!
此次没有报名的单位也不必惋惜,让小T哥先回来见见同事们,休整一下,给大家分享心得。下一次的巡回宣讲活动可一定不能再错过了哦。
下面我们赶紧来看看在宣讲过程中老师们都提出了什么问题,说不定就是您关心的点呢。
◆ ◆ ◆ ◆
阅微基因 2019
科研技术春季巡回宣讲QA汇总
◆ ◆ ◆ ◆
微生物&代谢组 联合分析
Q1:如果一个疾病的肠道微生物研究已经被很多人做过了怎么办?还能继续做吗?
肠道微生物已经被证明与非常多的疾病都有密切关系,这也说明了大部分疾病种类都已经做过了肠道微生物的测序研究。那么对于研究相同疾病方向的老师来说,如何在相同疾病的探索上实现研究新意呢?我们认为有四种方法:
1 提升样本量,使研究的数据更加饱满,能得到更加可信、真实的大数据结果;
2 关注功能,通过补充宏基因组测序,将菌群功能与菌群组成进一步结合并验证;
3 如果之前的研究没有关注代谢组学的话,可以加入代谢组的研究,进行微生物与代谢组学联合分析,从其他组学的层面深入阐述问题;
4 就疾病本身,在分组处理方式和实验设计上寻找新意。
Q2:微生物多样性的文章会相对来说比较容易发表吗?
微生物的研究现在是热点,关注度高,具有重要的研究意义以及临床的可应用性。相对于其他研究方向,人体微生物多样性的研究更受关注。但是科研本身是一个探究的过程,我们能做的是保证合理的实验设计、取样方法、样本量、实验技术和数据分析等,而实验结果是否符合预期、文章是否能顺利发表、最终能发表到几分,这是不能被保证的。然而,随着微生物研究的深入,微生物与疾病的关联性被证明,我们相信在保证实验设计没有问题的情况下,结果应当可以顺利发表。当然,除了单一组学,还要从多组学的角度上更加深入的阐述问题,甚至考虑进宿主基因对于微生物的互作影响。
Q3:做代谢组和微生物组的关联分析,取样上要注意什么?
微生物基因组和代谢组关联分析,如果是相同类型的样本,一定要取同一份后进行分装,一份用于微生物测序,另一份用于代谢组学检测。如果是不同种类样本尽量在同一时间段(12h内)取样,相隔太久取样无法关联。此外,如果涉及给药后影响,则需要考虑药物进入血液的时间和排泄的时间。

Q4:不同类型的样本用于测序和代谢组学的检测,是否能够关联?
同一个体上不同类型的样本进行微生物测序和代谢组学检测后是可以进行关联的。但是不同的联合方式研究的侧重点有所不同,好比下图举例的三种样本类型。此外,也可根据实验需求进行不同类型样本的组合。
Q5:肠道内容物与粪便样本检测有什么区别,该怎么样去选择?
粪样和肠道内容物都是常见的用于检测肠道微生物的样本类型。粪样可以粗略地认为是宿主整体肠道微生物的平均体现,取样方式方便,可操作性强;肠道内容物则需考虑肠道各部位的差异,例如大肠、小肠和盲肠部位的肠道微生物组成会有所不同,根据实验需求进行选择。
一般来说小肠主要负责食物的消化吸收,而大肠,尤其是微生物数量较多的盲肠,与微生物发酵有关。已有的一些研究通过比较粪样与不同肠段中的微生物,发现粪样与小肠的微生物谱不太相同,而与大肠较为相似。
Q6:代谢组学检测什么情况下选择非靶向广筛,什么情况下选择靶向检测?
当没有具体思路和明确的检测标的时,可以尝试先进行代谢组广筛(GC-MS或者LC-MS),以得到差异较大或者丰度高的代谢产物,进行下一步的实验计划。如有微生物测序结果,则可通过微生物的显著差异菌种或其对应的代谢通路分析,再选择进行靶向检测。
如果前人已有相关研究,或者自身具有研究基础,表明研究的疾病或者现象与某一代谢产物相关,则可以直接选择靶向检测。
Q7:神经递质的靶向检测好在哪里?
靶向检测相较于广筛方法的优势在于对目标产物(尤其是低浓度目标物质)的检测准确性。利用广筛方法进行神经递质的检测是有一定局限的,一是由于没有标准品,所以物质种类的鉴定结果是一个相似百分比。其二,广筛只能检测出浓度较高的神经递质,如血清素等,而一些浓度低或浓度梯度较大的神经递质,例如肾上腺素,则很难被检测出来,或检测出的浓度不准确。
Q8:不同16S区域的微生物测序是否可以兼容分析?
单就数据分析层面来说,不同区域(例如V4-V5区或V3-V4区)的测序结果可以通过比对数据库,得出菌种信息和丰度后将他们放在一起分析,因此技术上是可以兼容的。但当考虑到由于测序区域的不同,会导致数据库对比得出的菌种和丰度存在着一些差异,尤其是丰度较低的微生物类型,此时,便需要看是否把这个差异看做是一个较大的影响因素了。
Q9:做关联分析需要微生物测序与代谢组检测多少样本和生物学重复?
关联分析对每一个分组的生物学重复有要求,因为更多的样本数可以消除样本间的差异,提供更准确的代谢物检测结果,有助于提高代谢组与微生物基因组关联的结果质量与统计学意义。因此我们建议每个分组至少20-30对样本起(一对样本分别用于微生物测序和代谢组学检测)。
细胞鉴定
Q1:为什么小鼠细胞鉴定只能鉴定是否为小鼠细胞?而不能鉴定出种属?
人源细胞鉴定可以做到细胞株系的鉴定和是否有交叉污染现象,是基于将检测出的分型结果与ATCC或DSMZ等公开细胞库中的人源细胞STR分型数据进行比对,得到相关信息。而小鼠或其他物种的细胞STR信息在公共数据库中没有构建,所以检测结果无法比对,因此只能鉴定到是不是小鼠细胞和有没有交叉污染,不能鉴定到种属。
Q2:细胞鉴定图谱中,如何确定不均衡性?
在一个基因座上,杂合型的两个峰的面积比值大于2:1;或纯合型的单峰的值超过原本单峰面积的2倍时,可以判定此基因座存在不均衡性现象。
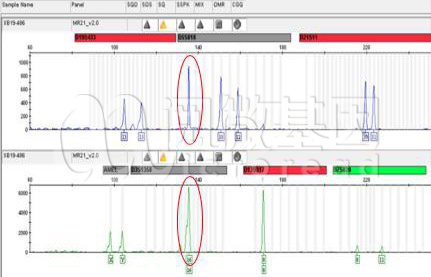
Q3:何为渗透峰,渗透峰的出现是否会影响分型的判断?
下图的蓝色通道中,红圈的峰就可以判定为一个典型的渗透峰,因为渗透峰的高度、位置、形状都和绿色通道中相同位置的峰非常相似。这是由于绿色通道基因座的信号过强,渗透到蓝色通道发生的。当渗透峰被清晰判断出来后,就可以对其进行排除,不会影响基因分型的判断。
SNP/SSR分子标记
Q1:一段基因中可能存在多种突变时,该如何进行检测?
一段基因当中存在多种形式的突变或多态性分子标记,例如SNP、SSR、InDel、CNV等。那么针对单一突变或者分子标记的检测方法是不通用的。建议依据片段长度进行一次或分段扩增测序。
Q2:如何确定筛选出来的SNP的分型,PIC值如何选择?
衡量DNA变异程度的重要指标可以用PIC值(多态性信息)来表示。当PIC值≥ 0.5 时表明其高度多态性,当0.25≤PIC值<0.5时, 表明其为中度多态性, 当PIC值< 0.25 时表明为低度多态性。
一般来说,实验中所得的SNP分子标记的PIC在0.3-0.4左右,这属于中度多态性。当然,除了PIC值之外,主等位基因频率、杂合度、基因多样性等参数也可作为参考。
Q3:用遗传分子标记筛选出来的和用表型筛选出来的不一致怎么办?
遗传分子标记只是一种品种鉴定的方法,虽然最终可以达到品种的区分和鉴定,但是所用的区分方法是分子层面上的。因此基于遗传分子标记筛选出的结果和表型筛选出的不一致,这种情况是时有发生的。因为单一分子层面上的分子标记可能不足以代表其表型特征。因此最好的方法是使用表型特征联合分子标记的方法进行品种鉴定。
Q4:进行动植物指纹图谱或品种鉴定研究的时候,SSR和SNP分别应该在什么情况下面进行选择?
一般来说,对于没有参考基因组,研究较少的物种来说SSR分子标记是比较合适的,因为SSR作为动植物的指纹图谱分子标记是被最广泛利用的,需要的位点少,且检测方法已被流程化、标准化。而相较于SSR来说,虽然SNP在基因组中密度高、代表性强、遗传稳定性好,但是通常需要更多位点(通常是SSR位点的4-5倍)和基因组信息才能达到目的,因此相对适合于研究较为成熟物种的指纹图谱或者分子身份证的构建。
Q5:进行SNP分子标记筛选的大概步骤是怎么样的?
大致可以分为三个步骤:
1 大量SNP位点的开发。可以通过重测序、简化基因组测序、基因芯片等方法获得;
2 利用不同品种的少量样本,基于主等位基因频率、杂合度、基因多样性和PIC值等参数,对多态性好的SNP位点进行筛选。选出最终的核心SNP位点;
3 用挑选出来的核心多态位点对大量不同品种的样本进行检测,验证核心位点区分不同物种的可行性。
Q6:分子标记开发都有哪些不同的方法?
详情见下表:
全转录组测序
Q1:全转录组测序有哪些建库的方法?
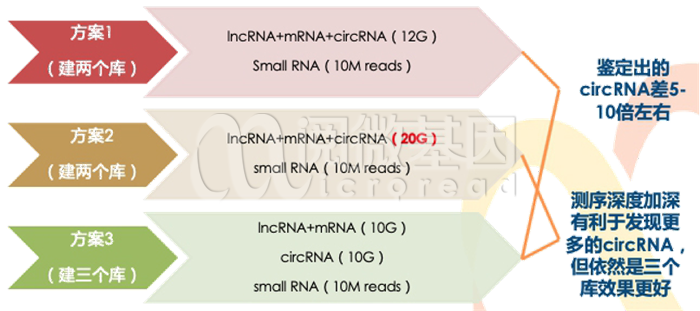
全转录组测序有三种建库方案,区别主要在于检出circRNA的数量。不同建库策略的成本也有所差别,具体参考下图。
Q2:全转录组测序后,在验证circRNA时的设计引物需要注意什么?
环状RNA在进行荧光定量验证时,是否能成功验证的关键点在于基于BSJ(反向剪切位点)设计的引物是否能特异性地扩增出环状RNA。环状RNA的引物设计要考虑到与BSJ点的距离、简并碱基和引物长度等。
甲基化检测
Q1:全基因组甲基化测序后如何选择位点进行检测?都有哪些合适的检测方法?
通常在进行过全基因组甲基化测序后,可对不同实验分组比较出显著差异的甲基化位点和其基因信息与功能。如果有原本就比较关注的功能基因,可以优先去测序数据中看这些基因的甲基化差异情况;如果没有关注的特定基因,可以通过对高通量甲基化检测进行整体筛选,然后通过数据分析挖掘出最显著差异的甲基化位点,并对挑选出来的甲基化位点对应的差异甲基化基因进行KEGG/GO功能分析,或综合进行性状关联分析,从而定位出核心甲基化位点基因。接下来可对挑选出来的核心基因的基因长度及甲基化位点分布情况等特征进行评估,以选择合适的方法,例如BSP、 MSP或焦磷酸进行下一步验证。

◆ ◆ ◆ ◆
宣讲风采
◆ ◆ ◆ ◆
以上就是在本次宣讲中精选出来的一些问题,有没有满足您心中的疑问呢?如果还有其他疑问,或者需要报名下一轮定制宣讲的话,请扫描文末的二维码关注公众号,留言联系我们。非常期待您的垂询呦。
相关产品详情请点击:
DNA甲基化检测
丁香通“科研伴我行”大促
为了答谢新老客户,阅微基因在2019.5.20-2019.6.9推出促销活动。详情请点击下方标题或首页活动海报。

细胞鉴定

微生物代谢组联合分析